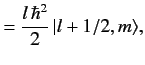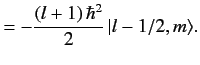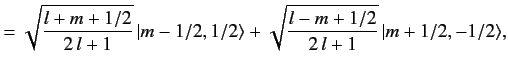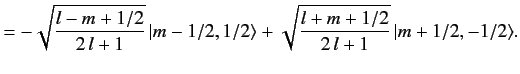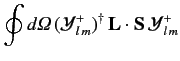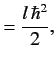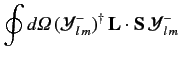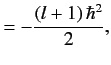

Next: Zeeman Effect
Up: Time-Independent Perturbation Theory
Previous: Linear Stark Effect
Fine Structure
Let us now consider the energy levels of hydrogen-like atoms (i.e., alkali
metal atoms) in more detail. The outermost electron moves in a spherically
symmetric potential 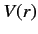 due to the nuclear charge and the charges of the
other electrons (which occupy spherically symmetric closed shells). The
shielding effect of the inner electrons causes
to depart from
the pure Coulomb form. This splits the degeneracy of states characterized by the
same value of
due to the nuclear charge and the charges of the
other electrons (which occupy spherically symmetric closed shells). The
shielding effect of the inner electrons causes
to depart from
the pure Coulomb form. This splits the degeneracy of states characterized by the
same value of 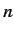 , but different values of
, but different values of 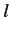 . In fact, higher
states
have higher energies.
. In fact, higher
states
have higher energies.
Let us examine a phenomenon known as fine structure, which is due to
interaction between the spin and orbital angular momenta of the outermost
electron. This electron experiences an electric field
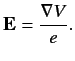 |
(679) |
However, a non-relativistic charge moving in an electric field also experiences an effective
magnetic field
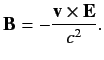 |
(680) |
Now, an electron possesses a spin magnetic moment [see Equation (462)]
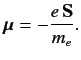 |
(681) |
We, therefore, expect a spin-orbit contribution to the Hamiltonian of
the form
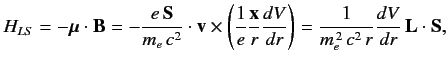 |
(682) |
where
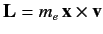 is the orbital angular momentum.
Actually, when the above expression is compared to the observed spin-orbit interaction,
it is found to be too large by a factor of two. There is a classical explanation
for this, due to spin precession, which we need not go into. The correct
quantum mechanical explanation requires a relativistically covariant
treatment of electron dynamics (this is achieved using the so-called Dirac equation--see Chapter 11).
is the orbital angular momentum.
Actually, when the above expression is compared to the observed spin-orbit interaction,
it is found to be too large by a factor of two. There is a classical explanation
for this, due to spin precession, which we need not go into. The correct
quantum mechanical explanation requires a relativistically covariant
treatment of electron dynamics (this is achieved using the so-called Dirac equation--see Chapter 11).
Let us now apply perturbation theory to a hydrogen-like atom, using 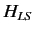 as the perturbation (with
taking one half of the value given above), and
as the perturbation (with
taking one half of the value given above), and
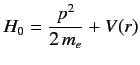 |
(683) |
as the unperturbed Hamiltonian. We have two choices for the energy
eigenstates of 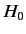 . We can adopt the simultaneous eigenstates of
. We can adopt the simultaneous eigenstates of
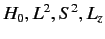 and
and 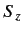 , or the simultaneous eigenstates of
, or the simultaneous eigenstates of
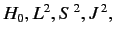 and
and 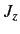 , where
, where
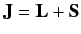 is
the total angular momentum. Although the departure of
from a pure
is
the total angular momentum. Although the departure of
from a pure
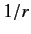 form splits the degeneracy of same
, different
, states,
those states characterized by the same values of
and
, but different
values of
form splits the degeneracy of same
, different
, states,
those states characterized by the same values of
and
, but different
values of 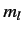 , are still degenerate.
(Here,
, are still degenerate.
(Here, 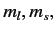 and
and 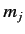 are the quantum numbers
corresponding to
are the quantum numbers
corresponding to 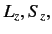 and
, respectively.)
Moreover, with the addition of spin
degrees of freedom, each state is doubly degenerate due to the two possible
orientations of the electron spin (i.e.,
and
, respectively.)
Moreover, with the addition of spin
degrees of freedom, each state is doubly degenerate due to the two possible
orientations of the electron spin (i.e.,
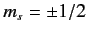 ). Thus, we are still
dealing with a
highly degenerate system. We know, from Section 7.6, that the application of
perturbation theory to a degenerate system is greatly simplified if the
basis eigenstates of the unperturbed Hamiltonian are also eigenstates
of the perturbing Hamiltonian. Now, the perturbing Hamiltonian,
, is proportional to
). Thus, we are still
dealing with a
highly degenerate system. We know, from Section 7.6, that the application of
perturbation theory to a degenerate system is greatly simplified if the
basis eigenstates of the unperturbed Hamiltonian are also eigenstates
of the perturbing Hamiltonian. Now, the perturbing Hamiltonian,
, is proportional to
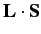 , where
, where
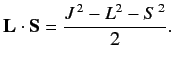 |
(684) |
It is fairly obvious
that the first group of operators (
and
)
does not commute with
, whereas the second group
(
and
) does. In fact,
is just a combination of operators appearing in the second group. Thus, it is
advantageous to work in terms of the eigenstates of the second group of
operators, rather than those of the first group.
We now need to find the simultaneous eigenstates of
and
.
This is equivalent to finding the eigenstates of the total angular momentum
resulting from the addition of two angular momenta: 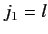 , and
, and
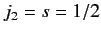 .
According to Equation (568), the allowed values of the total angular
momentum are
.
According to Equation (568), the allowed values of the total angular
momentum are 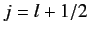 and
and 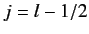 . We can write
. We can write
Here, the kets on the left-hand side are
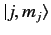 kets, whereas
those on the right-hand side are
kets, whereas
those on the right-hand side are
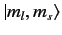 kets
(the
kets
(the 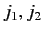 labels have been dropped, for the sake of clarity). We have made use
of the fact that the Clebsch-Gordon coefficients are automatically
zero unless
labels have been dropped, for the sake of clarity). We have made use
of the fact that the Clebsch-Gordon coefficients are automatically
zero unless
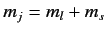 . We have also made use of the fact that
both the
and
kets are orthonormal,
and have unit lengths. We now need to determine
. We have also made use of the fact that
both the
and
kets are orthonormal,
and have unit lengths. We now need to determine
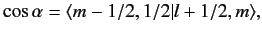 |
(687) |
where the Clebsch-Gordon coefficient is written in
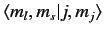 form.
form.
Let us now employ the recursion relation for Clebsch-Gordon coefficients, Equation (574),
with
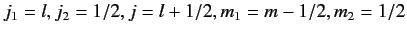 (lower sign).
We obtain
(lower sign).
We obtain
| |
![$\displaystyle [(l+1/2)\,(l+3/2)-m\,(m+1)]^{1/2} \,\langle m-1/2, 1/2\vert l+1/2, m\rangle$](img1643.png) |
|
| |
![$\displaystyle = [l\,(l+1)-(m-1/2)\,(m+1/2)]^{1/2}\, \langle m+1/2, 1/2\vert l+1/2, m+1\rangle,$](img1644.png) |
(688) |
which reduces to
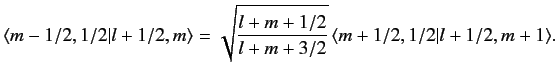 |
(689) |
We can use this formula to successively increase the value of
. For
instance,
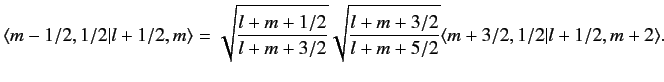 |
(690) |
This procedure can be continued until
attains its maximum possible value,
. Thus,
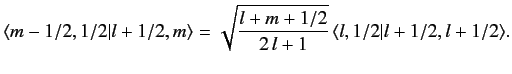 |
(691) |
Consider the situation in which
and 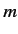 both take their maximum values,
and
both take their maximum values,
and 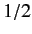 , respectively. The corresponding value of
is
, respectively. The corresponding value of
is
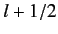 . This value is possible when
, but not when
.
Thus, the
ket
. This value is possible when
, but not when
.
Thus, the
ket
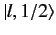 must be equal to
the
ket
must be equal to
the
ket
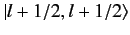 , up to an arbitrary phase-factor.
By convention, this factor is taken to be unity, giving
, up to an arbitrary phase-factor.
By convention, this factor is taken to be unity, giving
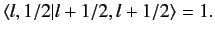 |
(692) |
It follows from Equation (691) that
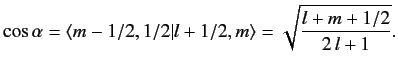 |
(693) |
Hence,
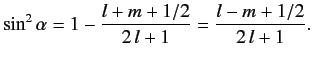 |
(694) |
We now need to determine the sign of
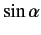 . A careful examination
of the recursion relation, Equation (574), shows that the plus sign is
appropriate. Thus,
. A careful examination
of the recursion relation, Equation (574), shows that the plus sign is
appropriate. Thus,
It is convenient to define so called spin-angular functions using the
Pauli two-component formalism:
These functions are eigenfunctions of the total angular momentum for spin
one-half particles, just as the spherical harmonics are eigenfunctions
of the orbital angular momentum. A general wavefunction for an energy
eigenstate in a hydrogen-like atom is written
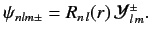 |
(698) |
The radial part of the wavefunction,
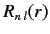 , depends on the radial
quantum number
and the angular quantum number
. The wavefunction
is also
labeled by
, which is the quantum number associated with
.
For a given choice of
, the quantum number
, depends on the radial
quantum number
and the angular quantum number
. The wavefunction
is also
labeled by
, which is the quantum number associated with
.
For a given choice of
, the quantum number 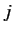 (i.e., the quantum number associated with
(i.e., the quantum number associated with 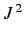 ) can take the values
) can take the values
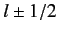 .
.
The
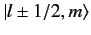 kets are eigenstates of
,
according to Equation (684).
Thus,
kets are eigenstates of
,
according to Equation (684).
Thus,
![$\displaystyle {\bf L} \cdot{\bf S}\, \vert j=l\pm 1/2,m_j= m\rangle = \frac{\hbar^2}{2} \left[ j\,(j+1) - l\,(l+1) - 3/4\right]\vert j,m\rangle,$](img1663.png) |
(699) |
giving
It follows that
where the integrals are over all solid angle,
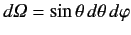 .
.
Let us now apply degenerate perturbation theory to evaluate the
shift in energy of a state whose wavefunction is
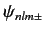 due to the spin-orbit Hamiltonian,
. To first order, the energy-shift is given by
due to the spin-orbit Hamiltonian,
. To first order, the energy-shift is given by
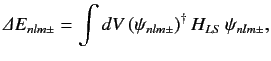 |
(704) |
where the integral is over all space,
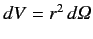 . Equations (682) (remembering the
factor of two), (698), and (702)-(703) yield
. Equations (682) (remembering the
factor of two), (698), and (702)-(703) yield
where
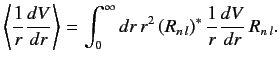 |
(707) |
Let us now apply the above result to the case of a sodium atom.
In chemist's notation, the ground state is written
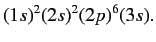 |
(708) |
The inner ten electrons effectively form a spherically symmetric electron
cloud. We are interested in the excitation of the eleventh electron
from 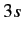 to some higher energy state. The closest (in energy) unoccupied
state is
to some higher energy state. The closest (in energy) unoccupied
state is 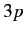 . This state has a higher energy than
due to the deviations
of the potential from the pure Coulomb form. In the absence of spin-orbit
interaction, there are six degenerate
states. The spin-orbit
interaction breaks the degeneracy of these states. The modified states are
labeled
. This state has a higher energy than
due to the deviations
of the potential from the pure Coulomb form. In the absence of spin-orbit
interaction, there are six degenerate
states. The spin-orbit
interaction breaks the degeneracy of these states. The modified states are
labeled
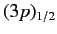 and
and
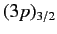 , where the subscript refers to the
value of
. The four
states lie at a slightly higher
energy level than the two
states,
because the radial integral (707) is positive. The splitting of
the
, where the subscript refers to the
value of
. The four
states lie at a slightly higher
energy level than the two
states,
because the radial integral (707) is positive. The splitting of
the 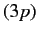 energy levels of the sodium atom can be observed
using a spectroscope.
The well-known sodium D line is associated with transitions between
the
and
states. The fact that there are two slightly different
energy levels (note that spin-orbit coupling does not split
the
energy levels) means that the sodium D line actually consists
of two very closely spaced spectroscopic lines. It is easily
demonstrated that the ratio of the typical spacing of
Balmer lines to the splitting
brought about by spin-orbit interaction is about
energy levels of the sodium atom can be observed
using a spectroscope.
The well-known sodium D line is associated with transitions between
the
and
states. The fact that there are two slightly different
energy levels (note that spin-orbit coupling does not split
the
energy levels) means that the sodium D line actually consists
of two very closely spaced spectroscopic lines. It is easily
demonstrated that the ratio of the typical spacing of
Balmer lines to the splitting
brought about by spin-orbit interaction is about
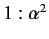 ,
where
,
where
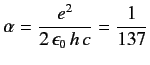 |
(709) |
is the fine structure constant. Actually, Equations (705)-(706) are not
entirely correct, because we have neglected an effect (namely, the
relativistic mass correction of the electron) that is the same
order of magnitude as spin-orbit coupling. (See Exercises 3 and 4.)
Next: Zeeman Effect
Up: Time-Independent Perturbation Theory
Previous: Linear Stark Effect
Richard Fitzpatrick
2013-04-08
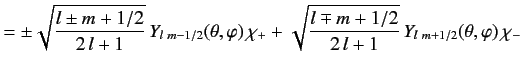
![$\displaystyle = \frac{1}{\sqrt{2\,l+1}}\left( \begin{array}{c} \pm \sqrt{l\pm m...
....5ex] \sqrt{l\mp m+1/2}\,\,Y_{l\,\,m+1/2}(\theta, \varphi) \end{array} \right).$](img1658.png)