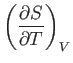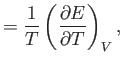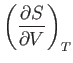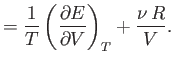

Next: Specific Heat
Up: Classical Thermodynamics
Previous: Introduction
Ideal Gas Equation of State
Let us commence our discussion of classical thermodynamics by considering the simplest possible macroscopic
system, which is an ideal gas. All of the thermodynamic properties of an ideal gas
are summed up in its equation of state, which specifies the relationship
between its pressure, volume, and temperature. Unfortunately, classical thermodynamics is unable determine this equation of state from first principles.
In fact, classical thermodynamics cannot determine anything from
first principles. We always need to provide some initial information
before classical thermodynamics can
generate new results.
This initial information may come from statistical physics (i.e., from our
knowledge of the microscopic structure of the system under
consideration), but, more usually, it is
empirical in nature
(i.e., it is obtained from experiments). In fact, the ideal gas
law was first discovered empirically by Robert Boyle. Nowadays, however, it
can be justified via statistical arguments, as is described in the following.
Recall
(from Section 3.8) that the number of accessible states of
a monotonic ideal gas varies like
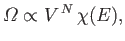 |
(6.6) |
where 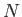 is the number of atoms, and
is the number of atoms, and 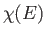 depends only on the energy
of the gas (and is, therefore,
independent of the volume). We obtained this result by integrating over the
volume of accessible phase-space. Because the energy of an ideal gas is independent of
the
atomic coordinates (given that there are no interatomic forces in an ideal gas), the integrals over these
coordinates just reduced to
simultaneous volume integrals, giving the
depends only on the energy
of the gas (and is, therefore,
independent of the volume). We obtained this result by integrating over the
volume of accessible phase-space. Because the energy of an ideal gas is independent of
the
atomic coordinates (given that there are no interatomic forces in an ideal gas), the integrals over these
coordinates just reduced to
simultaneous volume integrals, giving the
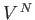 factor
in the previous expression. The integrals over the particle momenta were more
complicated, but were clearly independent of
factor
in the previous expression. The integrals over the particle momenta were more
complicated, but were clearly independent of 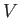 ,
giving the
factor in the previous expression.
Consider the following statistical result [see Equation (5.41)]:
,
giving the
factor in the previous expression.
Consider the following statistical result [see Equation (5.41)]:
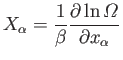 |
(6.7) |
where 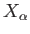 is the mean force conjugate to the external parameter
is the mean force conjugate to the external parameter 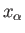 (i.e.,
(i.e.,
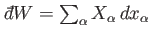 ), and
), and
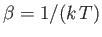 .
For
an ideal gas, the only external parameter is the volume, and its conjugate
force is the pressure (because
.
For
an ideal gas, the only external parameter is the volume, and its conjugate
force is the pressure (because
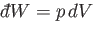 ). So, we can write
). So, we can write
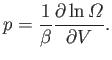 |
(6.8) |
It immediately follows from Equation (6.6) that
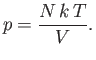 |
(6.9) |
However,
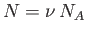 , where
, where 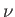 is the number of moles, and
is the number of moles, and 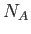 is Avagadro's
number. Also,
is Avagadro's
number. Also,
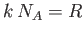 , where
, where 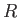 is the molar ideal gas constant. This allows us to
write the equation of state in its standard form
is the molar ideal gas constant. This allows us to
write the equation of state in its standard form
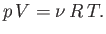 |
(6.10) |
Incidentally, the fact that
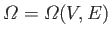 in Equation (6.6) suggests that the
macroscopic state of an ideal gas can be uniquely specified by giving the values of two independent parameters (e.g., the energy and the volume,
the pressure and the volume, the temperature and the volume, et cetera).
in Equation (6.6) suggests that the
macroscopic state of an ideal gas can be uniquely specified by giving the values of two independent parameters (e.g., the energy and the volume,
the pressure and the volume, the temperature and the volume, et cetera).
The previous derivation of the ideal gas equation of state
is rather elegant. It is certainly far easier to
obtain the equation of state in this manner
than to treat the atoms that make up the
gas as tiny billiard balls
continually
bouncing off the walls of a container. The latter derivation is difficult to
perform correctly, because it is necessary
to average over all possible directions of
atomic motion. (See Section 7.16.) It is clear, from the previous derivation, that the crucial element
needed to obtain the ideal gas equation of state is the absence of interatomic forces.
This automatically gives rise to a variation of the number of accessible states
with 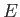 and
of
the form (6.6), which, in turn, implies the ideal gas law. So, the ideal gas law
should also apply to polyatomic gases with no inter-molecular forces. Polyatomic
gases are more complicated that monatomic gases because the molecules can rotate
and vibrate, giving rise to extra degrees of freedom, in addition to the
translational degrees of freedom of a monatomic gas. In other words,
, in Equation (6.6), becomes much more complicated
in polyatomic gases. However, as long as there
are no inter-molecular forces, the volume dependence of
and
of
the form (6.6), which, in turn, implies the ideal gas law. So, the ideal gas law
should also apply to polyatomic gases with no inter-molecular forces. Polyatomic
gases are more complicated that monatomic gases because the molecules can rotate
and vibrate, giving rise to extra degrees of freedom, in addition to the
translational degrees of freedom of a monatomic gas. In other words,
, in Equation (6.6), becomes much more complicated
in polyatomic gases. However, as long as there
are no inter-molecular forces, the volume dependence of
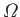 is still
, and the
ideal gas law should still hold true. In fact, we
shall discover that the extra degrees of freedom of polyatomic gases manifest
themselves by increasing the specific heat capacity. (See Section 7.13.)
is still
, and the
ideal gas law should still hold true. In fact, we
shall discover that the extra degrees of freedom of polyatomic gases manifest
themselves by increasing the specific heat capacity. (See Section 7.13.)
There is one other conclusion that we can draw from Equation (6.6). The statistical
definition of temperature is
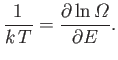 |
(6.11) |
[See Equation (5.31).]
It follows that
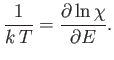 |
(6.12) |
We can see that, because 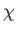 is a function of the energy, but not of the volume,
the temperature must also be a function of the
energy, but not the volume. This implies that
is a function of the energy, but not of the volume,
the temperature must also be a function of the
energy, but not the volume. This implies that
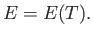 |
(6.13) |
In other words, the internal energy of an ideal gas depends only on the temperature
of the gas, and is independent of the volume.
This is a fairly obvious result, because if there are no inter-molecular forces
then increasing the volume, which
effectively increases the mean separation between molecules,
is not going to affect the molecular energies. Hence, the energy of the
whole gas is unaffected.
The volume independence of the internal energy can also
be derived directly from the ideal gas equation of state.
The internal energy of a gas can be considered to be a general function of the
temperature and volume, so that
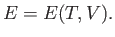 |
(6.14) |
It follows from mathematics that
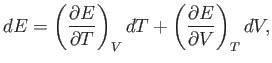 |
(6.15) |
where the subscript
reminds us that the first partial derivative is taken
at constant volume, and the subscript 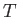 reminds us that the second
partial derivative
is taken at constant temperature. The first and second laws of thermodynamics imply that for a quasi-static
change of parameters,
reminds us that the second
partial derivative
is taken at constant temperature. The first and second laws of thermodynamics imply that for a quasi-static
change of parameters,
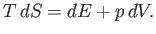 |
(6.16) |
[See Equation (6.5).]
The ideal gas equation of state, (6.10), can be used to express the pressure in term of the volume and
the temperature in the previous expression:
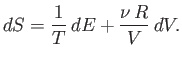 |
(6.17) |
Using Equation (6.15), this becomes
![$\displaystyle dS = \frac{1}{T} \left(\frac{\partial E}{\partial T}\right)_V dT ...
...{T} \left(\frac{\partial E}{\partial V}\right)_T + \frac{\nu R}{V}\right] dV.$](img877.png) |
(6.18) |
However, 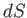 is the exact differential of a well-defined state function,
is the exact differential of a well-defined state function,
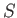 .
This means that we can consider the entropy to be a function of the temperature and
volume. Thus,
.
This means that we can consider the entropy to be a function of the temperature and
volume. Thus, 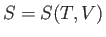 , and mathematics immediately yields
, and mathematics immediately yields
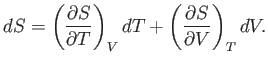 |
(6.19) |
The previous expression is valid for all small values of 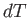 and
and 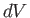 , so a comparison
with Equation (6.18) gives
, so a comparison
with Equation (6.18) gives
A well-known property of partial differentials is the equality of second
derivatives, irrespective of the order of differentiation, so
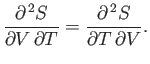 |
(6.22) |
This implies that
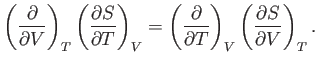 |
(6.23) |
The previous expression can be combined with Equations (6.20) and
(6.21) to give
![$\displaystyle \frac{1}{T} \left(\frac{\partial ^{ 2} E}{\partial V \partial ...
...ac{1}{T} \left (\frac{\partial^{ 2} E}{\partial T \partial V}\right)\right].$](img886.png) |
(6.24) |
Because second derivatives are equivalent, irrespective of the order of
differentiation,
the previous relation reduces to
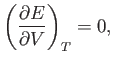 |
(6.25) |
which implies that the internal energy is independent of the volume for any gas obeying
the ideal equation of state. This result
was confirmed experimentally by James Joule in
the middle of the nineteenth century.
Next: Specific Heat
Up: Classical Thermodynamics
Previous: Introduction
Richard Fitzpatrick
2016-01-25